













设置登录密码
*密码
*确认密码
完善信息
*真实姓名
*公司名称
*您的职位
城市
*邮箱
*主营产品
*行业
企业类型
材质
功能







来源:中国科学院物理研究所|
发表时间:2019-01-31
点击:11148
磁性半金属(half metal)是一类重要的自旋电子学材料,具有非常独特的电子结构。在这类材料中,一种自旋取向(比如自旋朝上)的电子穿过费米面参与导电,形成金属特性;而另一种自旋取向(比如自旋朝下)的电子在费米面处打开能隙不参与导电,呈现绝缘体或半导体特性。因此,半金属的传导电子理论上具有100%的自旋极化率,可以提供完全自旋极化的载流子,被视为构建自旋电子器件的理想材料而备受关注。为使自旋电子器件能在常温下工作,半金属材料必须具有高于室温的(亚)铁磁居里温度。另一方面,为了提高传导电子的自旋极化率,需要尽可能抑制禁带热激活的跳跃电子,这就要求不参与导电的自旋取向的能带具有较宽的半金属能隙。如何从实验上设计并制备出具有高居里温度及宽半金属能隙(大于1.0 eV)的高性能磁性半金属新材料是极具挑战的科学问题。
近期,中国科学院物理研究所/北京凝聚态物理国家研究中心极端条件物理重点实验室EX6组龙有文研究员团队,利用独特的高温高压技术成功制备了同时具有A位与B位有序的多阶有序钙钛矿NaCu3Fe2Os2O12,发现该材料具有380 K的亚铁磁居里温度以及1.6 eV左右的半金属能隙,具有潜在的应用价值。
化学式为AA'3B2B'2O12的多阶有序钙钛矿是一种非常独特的结构载体,A'位、B位与B'位等三个原子位置可同时容纳过渡金属离子。因此,通过选取合适的离子组合,既可调节材料体系的磁相互作用,也可调节其电输运性质,为制备高性能半金属提供了可能。在前期同类结构的研究中,我们发现了首个具有立方钙钛矿结构的磁电多铁性材料(参见X. Wang et al., Phys. Rev. Lett. 115, 087601, 2015)、兼具大电极化和强磁电耦合效应的单相多功能材料(L. Zhou et al., Adv. Mater. 29, 1703435, 2017),以及居里温度高达580 K的新型亚铁磁半导体等(H. S. Deng et al, Phys. Rev. B 94, 024414, 2016)。最近,利用8-10 GPa与1200?C的高压高温实验条件,我们率先获得了一种新型多阶有序钙钛矿化合物NaCu3Fe2Os2O12。通过磁化率、磁化强度、电阻、磁电阻、同步辐射X光吸收谱(XAS)与磁圆二色谱(XMCD)等一系列综合结构表征与物性测试,并结合第一性原理理论计算,我们对该体系进行了详细研究。测试表明,该材料电荷组态为Cu2+/Fe3+/Os5.5+,并且这三种过渡金属离子均参与磁相互作用,形成Cu2+(↑)Fe3+(↑)Os5.5+(↓)的长程亚铁磁相互作用,居里温度为380 K,每个分子式的饱和磁矩为5.5μB。电输运测试表明该材料具有半金属类似的电阻-温度依赖关系,并展示了自旋阀型(spin-valve-type)的磁电阻现象,预示其自旋极化载流子可能源于晶粒间的隧穿效应,这些现象跟经典的磁性半金属材料Sr2FeMoO6非常类似。进一步,我们与极端条件实验室杨义峰研究员团队合作,计算了NaCu3Fe2Os2O12的电子结构,发现Cu和Fe的态密度远离费米面,而Os自旋朝下的电子穿过费米面,形成金属导电能带结构,但Os自旋朝上的电子则打开能隙,形成绝缘体特征,表明了NaCu3Fe2Os2O12的半金属特性。更有意思的是,计算发现该体系具有高达1.6 eV左右的半金属能隙,在目前所发现的半金属材料中处于最大水平,并且该能隙大小基本不随电子关联能(针对Cu、Fe、Os)与自旋-轨道耦合(针对Os)作用大小的变化而改变。因而,NaCu3Fe2Os2O12提供了一个具有室温以上亚铁磁居里温度以及宽能隙的高性能半金属,为自旋电子学器件的应用提供了重要候选材料。
相关研究结果发表在近期的Inorg. Chem. 58, 320(2019)上, 并被选为当期的封面。该工作获得了国家自然科学基金委(51772324, 11574378,11522435)、科技部(2018YFA0305700,2015CB921303)、中国科学院(YZ201555, GJHZ1773, XDB07000000,QYZDB-SSW-SLH013)等项目的支持。

图1:本工作被选为Inorg. Chem. 封面论文。
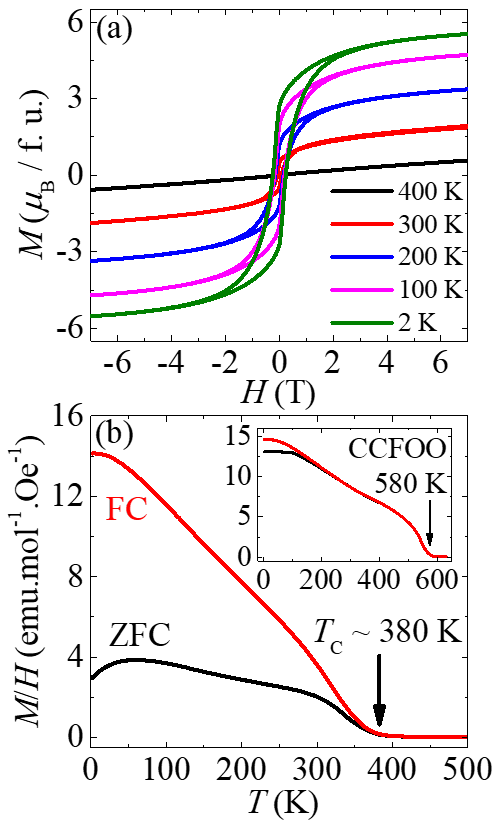
图2:NaCu 3 Fe 2 Os 2 O 12 的高温亚铁磁行为。
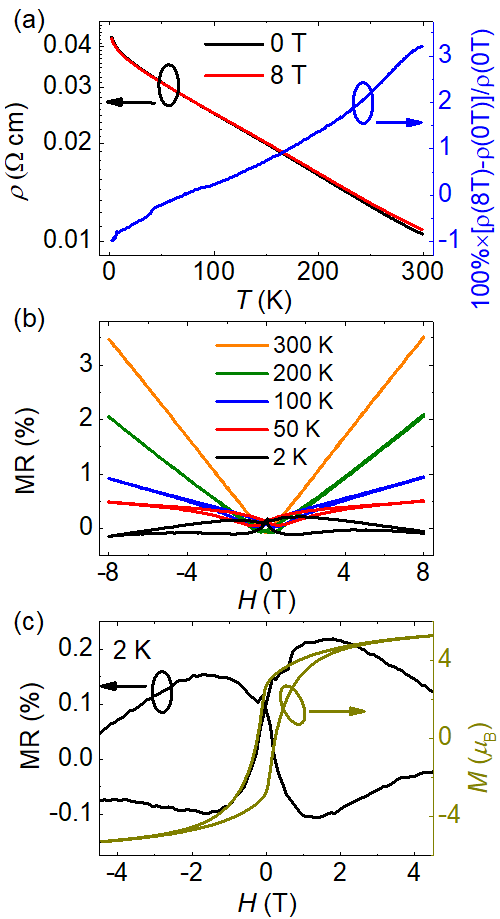
图3:NaCu 3 Fe 2 Os 2 O 12 的电输运测试结果。

图4:NaCu 3 Fe 2 Os 2 O 12 的第一性原理计算结果。
[声明]本文来源于互联网转载,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性、准确性等负责,尤其不对文中产品有关功能性、效果等提供担保。本站文章版权归原作者所有,内容为作者个人观点,本站提醒读者,文章仅供学习参考,不构成任何投资及应用建议,如需转载,请联系原作者。如涉及作品内容、版权和其它问题,请与我们联系,我们将在第一时间处理!本站拥有对此声明的最终解释权。





