













设置登录密码
*密码
*确认密码
完善信息
*真实姓名
*公司名称
*您的职位
城市
*邮箱
*主营产品
*行业
企业类型
材质
功能







来源:材料科技在线|
发表时间:2018-07-08
点击:14202
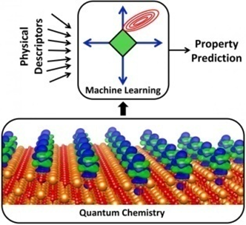
图片描绘了量子化学模拟金属原子和载体(下方橙色部分所示)之间的电荷转移(蓝色/绿色)现象,这只是对催化剂物理行为的一种描述。研究人员通过计算330000个催化剂这样的物理行为创建了一个庞大的数据库。通过机器智能学习(上图)在数据库中搜索隐藏的图案,设计师可使用这些图案来制造成本更低、效果更佳的催化剂。
研究人员研发出一种结合机器智能学习和量子化学的新方法,可使催化剂设计速度更快、制造成本更低。
莱斯大学和宾夕法尼亚州立大学的一个团队发现:机器智能学习和量子化学隐藏的相关性可以改善设计过程,科学家便可丢弃既耗时又昂贵的传统实验室测试手段,找到用于制备催化剂最佳的金属及其材料。
“在计算催化中会产生大量的数据,并且我们开始认识到在该领域,对于筛选高容量数据,以及寻找我们可能会错过的基本相关性,数据科学的工具就变得非常有价值,”Thomas Senftle——莱斯大学化学和生物分子工程助理教授在一份声明中这样说道。“这就是本文的真正含义。我们结合了成熟的数据生成和分析工具,使我们能够寻找我们原本不会注意到的相关性。”
研究人员首先利用密度泛函理论计算了几种不同类型金属单个原子的结合强度,以及一系列金属氧化物基底。
宾夕法尼亚州立大学化学工程教授Michael Janik说:“金属和基底之间的结合能极为引人关注,因为键越牢固,金属原子就越不容易被移除。如果我们能够控制这种结合能,我们就能调整这些金属粒子的大小分布。而这反过来又会影响它们所能催化的整体反应。
该小组为每个金属基底组合编排了大约330,000个附加属性,其中包括氧化物形成能量、配位数、合金生成自由能和电离能等因素。
论文的主要作者,宾夕法尼亚州立大学研究生研究助理A.S.M.Jonayat说:“机器智能学习算法的目的,是寻找那些与所观察到的结合能数据相关的描述符组合。我们便会思考这样一个问题:‘在所有这些描述符中,我们怎么才能找到那些我们真正感兴趣的描述符呢?’”
根据研究人员的说法,相关性的识别可以简化催化剂的设计,使其在耗时和昂贵的实验室测试之前便可对材料行为进行预测。
该团队下一步的计划是构建复杂性的模拟。“在这里,我们看到的是没有水分子或任何杂质的情况下金属和支撑物之间的相互作用,”Senftle说道。“在现实中,催化剂应用于非常复杂的反应环境中,我们想要研究这些趋势在这些环境中是如何变化的。”
“本文由新材料在线®平台入驻媒体号材料科技在线提供,观点仅代表作者本人,不代表本网站及新材料在线®立场,本站不对文章内容真实性、准确性等负责,尤其不对文中产品有关功能性、效果等提供担保。本站提醒读者,文章仅供学习参考,不构成任何投资及应用建议。如需转载,请联系原作者。如涉及作品内容、版权和其它问题,请与我们联系,我们将在第一时间处理!本站拥有对此声明的最终解释权。”





